 日本語
日本語 繁體中文
繁體中文 简体中文
简体中文 English
English
Publications
Selected Publications

High-resolution CMOS MEA platform to study neurons at subcellular, cellular, and network levels

Presenting measurements of neuronal preparations with a novel CMOS-based microelectrode array at high-spatiotemporal-resolution on subcellular, cellular, and network level.
J. Müller, M. Ballini, P. Livi, Y. Chen, M. Radivojevic, A. Shadmani, V. Viswam, I. L. Jones, M. Fiscella, R. Diggelmann, A. Stettler, U. Frey, D. J. Bakkum, and A. Hierlemann, “High-resolution CMOS MEA platform to study neurons at subcellular, cellular, and network levels,” Lab Chip, vol. 15, no. 13, pp. 2767–2780, May 2015.
Revealing Neuronal Function through Microelectrode Array Recordings

Reviewing the current understanding of microelectrode signals and the techniques for analyzing them, with focus on the ongoing advancements in microelectrode technology (in vivo and in vitro) and recent advanced microelectrode array measurement methods that facilitate the understanding of single neurons and network function.
M. E. J. Obien, K. Deligkaris, T. Bullmann, D. J. Bakkum, and U. Frey, “Revealing Neuronal Function through Microelectrode Array Recordings,” Front. Neurosci., 8:423, Jan 2015.

A 1024-Channel CMOS Microelectrode Array With 26,400 Electrodes for Recording and Stimulation of Electrogenic Cells In Vitro
A high-resolution CMOS-based microelectrode array featuring 1,024 low-noise readout channels, 26,400 electrodes at a density of 3,265 electrodes per mm2, including on-chip 10bit ADCs and consuming only 75 mW.
M. Ballini, J. Muller, P. Livi, Y. Chen, U. Frey, A. Stettler, A. Shadmani, V. Viswam, I. L. Jones, D. Jackel, M. Radivojevic, M. K. Lewandowska, W. Gong, M. Fiscella, D. J. Bakkum, F. Heer, and A. Hierlemann, “A 1024-Channel CMOS Microelectrode Array With 26,400 Electrodes for Recording and Stimulation of Electrogenic Cells In Vitro,” IEEE Journal of Solid-State Circuits, vol. 49, no. 11, pp. 2705-2719, 2014.
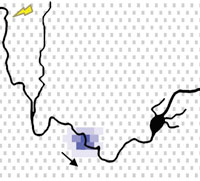
Tracking axonal action potential propagation on a high-density microelectrode array across hundreds of sites
Demonstrating a method to electrically visualize action potential propagation on axons and revealing
large variations in velocity.
D. J. Bakkum, U. Frey, M. Radivojevic, T. L. Russell, J. Muller, M. Fiscella, H. Takahashi, and A. Hierlemann, “Tracking axonal action potential propagation on a high-density microelectrode array across hundreds of sites,” Nature Communications, 4:2181, Jul 2013.

Microelectronic System for High-Resolution Mapping of Extracellular Electric Fields Applied to Brain Slices
Recording and modeling extracellular action potentials of Purkinje cells at subcellular resolution.
U. Frey, U. Egert, F. Heer, S. Hafizovic, and A. Hierlemann, “Microelectronic System for High-Resolution Mapping of Extracellular Electric Fields Applied to Brain Slices,” Biosensors and Bioelectronics, vol. 24, no. 7, pp. 2191-2198, 2009.

Modulation of Cardiomyocyte Electrical Properties Using Regulated Bone Morphogenetic Protein-2 Expression
Controlling BMP-2 expression to modulate the electrophysiological properties of cardiomyocytes using an HD-MEA for detailed monitoring.
C. D. Sanchez-Bustamante, U. Frey, J. M. Kelm, A. Hierlemann, and M. Fussenegger,
“Modulation of Cardiomyocyte Electrical Properties Using Regulated Bone Morphogenetic Protein-2 Expression,” Tissue Engineering Part A, vol. 14, no. 12, pp. 1969-1988, 2008.
All Publications
| 1. | Hruska-Plochan, Marian; Wiersma, Vera I; Betz, Katharina M; Mallona, Izaskun; Ronchi, Silvia; Maniecka, Zuzanna; Hock, Eva-Maria; Tantardini, Elena; Laferriere, Florent; Sahadevan, Sonu; Hoop, Vanessa; Delvendahl, Igor; Pérez-Berlanga, Manuela; Gatta, Beatrice; Panatta, Martina; van der Bourg, Alexander; Bohaciakova, Dasa; Sharma, Puneet; Vos, Laura De; Frontzek, Karl; Aguzzi, Adriano; Lashley, Tammaryn; Robinson, Mark D; Karayannis, Theofanis; Mueller, Martin; Hierlemann, Andreas; Polymenidou, Magdalini: A model of human neural networks reveals NPTX2 pathology in ALS and FTLD. In: Nature, 2024. (Type: Journal Article | Abstract | Links | BibTeX) @article{Hruska-Plochan2024, title = {A model of human neural networks reveals NPTX2 pathology in ALS and FTLD}, author = {Marian Hruska-Plochan and Vera I. Wiersma and Katharina M. Betz and Izaskun Mallona and Silvia Ronchi and Zuzanna Maniecka and Eva-Maria Hock and Elena Tantardini and Florent Laferriere and Sonu Sahadevan and Vanessa Hoop and Igor Delvendahl and Manuela Pérez-Berlanga and Beatrice Gatta and Martina Panatta and Alexander van der Bourg and Dasa Bohaciakova and Puneet Sharma and Laura De Vos and Karl Frontzek and Adriano Aguzzi and Tammaryn Lashley and Mark D. Robinson and Theofanis Karayannis and Martin Mueller and Andreas Hierlemann and Magdalini Polymenidou }, url = {https://www.nature.com/articles/s41586-024-07042-7}, doi = {10.1038/s41586-024-07042-7}, year = {2024}, date = {2024-02-14}, journal = {Nature}, abstract = {Human cellular models of neurodegeneration require reproducibility and longevity, which is necessary for simulating age-dependent diseases. Such systems are particularly needed for TDP-43 proteinopathies1, which involve human-specific mechanisms that cannot be directly studied in animal models. Here, to explore the emergence and consequences of TDP-43 pathologies, we generated induced pluripotent stem cell-derived, colony morphology neural stem cells (iCoMoNSCs) via manual selection of neural precursors. Single-cell transcriptomics and comparison to independent neural stem cells showed that iCoMoNSCs are uniquely homogenous and self-renewing. Differentiated iCoMoNSCs formed a self-organized multicellular system consisting of synaptically connected and electrophysiologically active neurons, which matured into long-lived functional networks (which we designate iNets). Neuronal and glial maturation in iNets was similar to that of cortical organoids. Overexpression of wild-type TDP-43 in a minority of neurons within iNets led to progressive fragmentation and aggregation of the protein, resulting in a partial loss of function and neurotoxicity. Single-cell transcriptomics revealed a novel set of misregulated RNA targets in TDP-43-overexpressing neurons and in patients with TDP-43 proteinopathies exhibiting a loss of nuclear TDP-43. The strongest misregulated target encoded the synaptic protein NPTX2, the levels of which are controlled by TDP-43 binding on its 3′ untranslated region. When NPTX2 was overexpressed in iNets, it exhibited neurotoxicity, whereas correcting NPTX2 misregulation partially rescued neurons from TDP-43-induced neurodegeneration. Notably, NPTX2 was consistently misaccumulated in neurons from patients with amyotrophic lateral sclerosis and frontotemporal lobar degeneration with TDP-43 pathology. Our work directly links TDP-43 misregulation and NPTX2 accumulation, thereby revealing a TDP-43-dependent pathway of neurotoxicity.}, keywords = {}, pubstate = {published}, tppubtype = {article} } Human cellular models of neurodegeneration require reproducibility and longevity, which is necessary for simulating age-dependent diseases. Such systems are particularly needed for TDP-43 proteinopathies1, which involve human-specific mechanisms that cannot be directly studied in animal models. Here, to explore the emergence and consequences of TDP-43 pathologies, we generated induced pluripotent stem cell-derived, colony morphology neural stem cells (iCoMoNSCs) via manual selection of neural precursors. Single-cell transcriptomics and comparison to independent neural stem cells showed that iCoMoNSCs are uniquely homogenous and self-renewing. Differentiated iCoMoNSCs formed a self-organized multicellular system consisting of synaptically connected and electrophysiologically active neurons, which matured into long-lived functional networks (which we designate iNets). Neuronal and glial maturation in iNets was similar to that of cortical organoids. Overexpression of wild-type TDP-43 in a minority of neurons within iNets led to progressive fragmentation and aggregation of the protein, resulting in a partial loss of function and neurotoxicity. Single-cell transcriptomics revealed a novel set of misregulated RNA targets in TDP-43-overexpressing neurons and in patients with TDP-43 proteinopathies exhibiting a loss of nuclear TDP-43. The strongest misregulated target encoded the synaptic protein NPTX2, the levels of which are controlled by TDP-43 binding on its 3′ untranslated region. When NPTX2 was overexpressed in iNets, it exhibited neurotoxicity, whereas correcting NPTX2 misregulation partially rescued neurons from TDP-43-induced neurodegeneration. Notably, NPTX2 was consistently misaccumulated in neurons from patients with amyotrophic lateral sclerosis and frontotemporal lobar degeneration with TDP-43 pathology. Our work directly links TDP-43 misregulation and NPTX2 accumulation, thereby revealing a TDP-43-dependent pathway of neurotoxicity. |
| 2. | Ryu, Seungmi; Weber, Claire; Chu, Pei-Hsuan; Ernest, Ben; Jovanovic, Vukasin M; Deng, Tao; Slamecka, Jaroslav; Hong, Hyenjong; Jethmalani, Yogita; Baskir, Hannah M; Inman, Jason; Braisted, John; Hirst, Marissa B; Simeonov, Anton; Voss, Ty C; Tristan, Carlos A; Singeç, Ilyas: Stress-free cell aggregation by using the CEPT cocktail enhances embryoid body and organoid fitness. In: Biofabrication, 2023. (Type: Journal Article | Abstract | Links | BibTeX) @article{Ryu2023, title = {Stress-free cell aggregation by using the CEPT cocktail enhances embryoid body and organoid fitness}, author = {Seungmi Ryu and Claire Weber and Pei-Hsuan Chu and Ben Ernest and Vukasin M Jovanovic and Tao Deng and Jaroslav Slamecka and Hyenjong Hong and Yogita Jethmalani and Hannah M Baskir and Jason Inman and John Braisted and Marissa B Hirst and Anton Simeonov and Ty C Voss and Carlos A Tristan and Ilyas Singeç}, url = {https://dx.doi.org/10.1088/1758-5090/ad0d13}, doi = {10.1088/1758-5090/ad0d13}, year = {2023}, date = {2023-12-11}, journal = {Biofabrication}, abstract = {Embryoid bodies (EBs) and self-organizing organoids derived from human pluripotent stem cells (hPSCs) recapitulate tissue development in a dish and hold great promise for disease modeling and drug development. However, current protocols are hampered by cellular stress and apoptosis during cell aggregation, resulting in variability and impaired cell differentiation. Here, we demonstrate that EBs and various organoid models (e.g., brain, gut, kidney) can be optimized by using the small molecule cocktail named CEPT (chroman 1, emricasan, polyamines, trans-ISRIB), a polypharmacological approach that ensures cytoprotection and cell survival. Application of CEPT for just 24 h during cell aggregation has long-lasting consequences affecting morphogenesis, gene expression, cellular differentiation, and organoid function. Various qualification methods confirmed that CEPT treatment enhanced experimental reproducibility and consistently improved EB and organoid fitness as compared to the widely used ROCK inhibitor Y-27632. Collectively, we discovered that stress-free cell aggregation and superior cell survival in the presence of CEPT are critical quality control determinants that establish a robust foundation for bioengineering complex tissue and organ models.}, keywords = {}, pubstate = {published}, tppubtype = {article} } Embryoid bodies (EBs) and self-organizing organoids derived from human pluripotent stem cells (hPSCs) recapitulate tissue development in a dish and hold great promise for disease modeling and drug development. However, current protocols are hampered by cellular stress and apoptosis during cell aggregation, resulting in variability and impaired cell differentiation. Here, we demonstrate that EBs and various organoid models (e.g., brain, gut, kidney) can be optimized by using the small molecule cocktail named CEPT (chroman 1, emricasan, polyamines, trans-ISRIB), a polypharmacological approach that ensures cytoprotection and cell survival. Application of CEPT for just 24 h during cell aggregation has long-lasting consequences affecting morphogenesis, gene expression, cellular differentiation, and organoid function. Various qualification methods confirmed that CEPT treatment enhanced experimental reproducibility and consistently improved EB and organoid fitness as compared to the widely used ROCK inhibitor Y-27632. Collectively, we discovered that stress-free cell aggregation and superior cell survival in the presence of CEPT are critical quality control determinants that establish a robust foundation for bioengineering complex tissue and organ models. |
| 3. | Elliott, Matthew A T; Schweiger, Hunter E; Robbins, Ash; Vera-Choqqueccota, Samira; Ehrlich, Drew; Hernandez, Sebastian; Voitiuk, Kateryna; Geng, Jinghui; Sevetson, Jess L; Core, Cordero; Rosen, Yohei M; Teodorescu, Mircea; Wagner, Nico O; Haussler, David; Mostajo-Radji, Mohammed A: Internet-Connected Cortical Organoids for Project-Based Stem Cell and Neuroscience Education. In: eNeuro, 2023. (Type: Journal Article | Abstract | Links | BibTeX) @article{Elliott2023, title = {Internet-Connected Cortical Organoids for Project-Based Stem Cell and Neuroscience Education}, author = {Matthew A. T. Elliott and Hunter E. Schweiger and Ash Robbins and Samira Vera-Choqqueccota and Drew Ehrlich and Sebastian Hernandez and Kateryna Voitiuk and Jinghui Geng and Jess L. Sevetson and Cordero Core and Yohei M. Rosen and Mircea Teodorescu and Nico O. Wagner and David Haussler and Mohammed A. Mostajo-Radji}, url = {https://www.eneuro.org/lookup/doi/10.1523/ENEURO.0308-23.2023}, doi = {10.1523/ENEURO.0308-23.2023}, year = {2023}, date = {2023-11-28}, journal = {eNeuro}, abstract = {The introduction of Internet-connected technologies to the classroom has the potential to revolutionize STEM education by allowing students to perform experiments in complex models that are unattainable in traditional teaching laboratories. By connecting laboratory equipment to the cloud, we introduce students to experimentation in pluripotent stem cell (PSC)-derived cortical organoids in two different settings: using microscopy to monitor organoid growth in an introductory tissue culture course and using high-density (HD) multielectrode arrays (MEAs) to perform neuronal stimulation and recording in an advanced neuroscience mathematics course. We demonstrate that this approach develops interest in stem cell and neuroscience in the students of both courses. All together, we propose cloud technologies as an effective and scalable approach for complex project-based university training.}, keywords = {}, pubstate = {published}, tppubtype = {article} } The introduction of Internet-connected technologies to the classroom has the potential to revolutionize STEM education by allowing students to perform experiments in complex models that are unattainable in traditional teaching laboratories. By connecting laboratory equipment to the cloud, we introduce students to experimentation in pluripotent stem cell (PSC)-derived cortical organoids in two different settings: using microscopy to monitor organoid growth in an introductory tissue culture course and using high-density (HD) multielectrode arrays (MEAs) to perform neuronal stimulation and recording in an advanced neuroscience mathematics course. We demonstrate that this approach develops interest in stem cell and neuroscience in the students of both courses. All together, we propose cloud technologies as an effective and scalable approach for complex project-based university training. |
| 4. | Sawada, Tomoyo; Barbosa, André R; Araujo, Bruno; McCord, Alejandra E; D’Ignazio, Laura; Benjamin, Kynon J M; Sheehan, Bonna; Zabolocki, Michael; Feltrin, Arthur; Arora, Ria; Brandtjen, Anna C; Kleinman, Joel E; Hyde, Thomas M; Bardy, Cedric; Weinberger, Daniel R; Paquola, Apuã C M; Erwin, Jennifer A: Recapitulation of Perturbed Striatal Gene Expression Dynamics of Donor’s Brains With Ventral Forebrain Organoids Derived From the Same Individuals With Schizophrenia. In: American Journal of Psychiatry, 2023, ISSN: 0002-953X. (Type: Journal Article | Abstract | Links | BibTeX) @article{Tomoyo2023, title = {Recapitulation of Perturbed Striatal Gene Expression Dynamics of Donor’s Brains With Ventral Forebrain Organoids Derived From the Same Individuals With Schizophrenia}, author = {Tomoyo Sawada and André R. Barbosa and Bruno Araujo and Alejandra E. McCord and Laura D’Ignazio and Kynon J.M. Benjamin and Bonna Sheehan and Michael Zabolocki and Arthur Feltrin and Ria Arora and Anna C. Brandtjen and Joel E. Kleinman and Thomas M. Hyde and Cedric Bardy and Daniel R. Weinberger and Apuã C.M. Paquola and Jennifer A. Erwin}, url = {https://ajp.psychiatryonline.org/doi/10.1176/appi.ajp.20220723}, doi = {10.1176/appi.ajp.20220723}, issn = {0002-953X}, year = {2023}, date = {2023-11-02}, journal = {American Journal of Psychiatry}, abstract = {Objective: Schizophrenia is a brain disorder that originates during neurodevelopment and has complex genetic and environmental etiologies. Despite decades of clinical evidence of altered striatal function in affected patients, studies examining its cellular and molecular mechanisms in humans are limited. To explore neurodevelopmental alterations in the striatum associated with schizophrenia, the authors established a method for the differentiation of induced pluripotent stem cells (iPSCs) into ventral forebrain organoids (VFOs). Methods: VFOs were generated from postmortem dural fibroblast–derived iPSCs of four individuals with schizophrenia and four neurotypical control individuals for whom postmortem caudate genotypes and transcriptomic data were profiled in the BrainSeq neurogenomics consortium. Individuals were selected such that the two groups had nonoverlapping schizophrenia polygenic risk scores (PRSs). Results: Single-cell RNA sequencing analyses of VFOs revealed differences in developmental trajectory between schizophrenia and control individuals in which inhibitory neuronal cells from the patients exhibited accelerated maturation. Furthermore, upregulated genes in inhibitory neurons in schizophrenia VFOs showed a significant overlap with upregulated genes in postmortem caudate tissue of individuals with schizophrenia compared with control individuals, including the donors of the iPSC cohort. Conclusions: The findings suggest that striatal neurons derived from high-PRS individuals with schizophrenia carry abnormalities that originated during early brain development and that the VFO model can recapitulate disease-relevant cell type–specific neurodevelopmental phenotypes in a dish.}, keywords = {}, pubstate = {published}, tppubtype = {article} } Objective: Schizophrenia is a brain disorder that originates during neurodevelopment and has complex genetic and environmental etiologies. Despite decades of clinical evidence of altered striatal function in affected patients, studies examining its cellular and molecular mechanisms in humans are limited. To explore neurodevelopmental alterations in the striatum associated with schizophrenia, the authors established a method for the differentiation of induced pluripotent stem cells (iPSCs) into ventral forebrain organoids (VFOs). Methods: VFOs were generated from postmortem dural fibroblast–derived iPSCs of four individuals with schizophrenia and four neurotypical control individuals for whom postmortem caudate genotypes and transcriptomic data were profiled in the BrainSeq neurogenomics consortium. Individuals were selected such that the two groups had nonoverlapping schizophrenia polygenic risk scores (PRSs). Results: Single-cell RNA sequencing analyses of VFOs revealed differences in developmental trajectory between schizophrenia and control individuals in which inhibitory neuronal cells from the patients exhibited accelerated maturation. Furthermore, upregulated genes in inhibitory neurons in schizophrenia VFOs showed a significant overlap with upregulated genes in postmortem caudate tissue of individuals with schizophrenia compared with control individuals, including the donors of the iPSC cohort. Conclusions: The findings suggest that striatal neurons derived from high-PRS individuals with schizophrenia carry abnormalities that originated during early brain development and that the VFO model can recapitulate disease-relevant cell type–specific neurodevelopmental phenotypes in a dish. |
| 5. | Kelley, Matt R; Chipman, Laura B; Asano, Shoh; Knott, Matthew; Howard, Samantha T; Berg, Allison P: Potentiating NaV1.1 in Dravet syndrome patient iPSC-derived GABAergic neurons increases neuronal firing frequency and decreases network synchrony. In: bioRxiv, 2023. (Type: Journal Article | Abstract | Links | BibTeX) @article{Kelley2023, title = {Potentiating NaV1.1 in Dravet syndrome patient iPSC-derived GABAergic neurons increases neuronal firing frequency and decreases network synchrony}, author = {Matt R Kelley and Laura B Chipman and Shoh Asano and Matthew Knott and Samantha T Howard and Allison P Berg}, url = {https://www.biorxiv.org/content/10.1101/2023.09.28.559990v1}, doi = {10.1101/2023.09.28.559990}, year = {2023}, date = {2023-09-29}, journal = {bioRxiv}, abstract = {Dravet syndrome is a developmental and epileptic encephalopathy characterized by seizures, behavioral abnormalities, developmental deficits, and elevated risk of sudden unexpected death in epilepsy (SUDEP). Most patient cases are caused by de novo loss-of-function mutations in the gene SCN1A, causing a haploinsufficiency of the alpha subunit of the voltage-gated sodium channel NaV1.1. Within the brain, NaV1.1 is primarily localized to the axons of inhibitory neurons, and decreased NaV1.1 function is hypothesized to reduce GABAergic inhibitory neurotransmission within the brain, driving neuronal network hyperexcitability and subsequent pathology. We have developed a human in vitro model of Dravet syndrome using differentiated neurons derived from patient iPSC and enriched for GABA expressing neurons. Neurons were plated on high definition multielectrode arrays (HD-MEAs), permitting recordings from the same cultures over the 7-weeks duration of study at the network, single cell, and subcellular resolution. Using this capability, we characterized the features of axonal morphology and physiology. Neurons developed increased spiking activity and synchronous network bursting. Recordings were processed through a spike sorting pipeline for curation of single unit activity and to assess the effects of pharmacological treatments. At 7-weeks, the application of the GABAAR receptor agonist muscimol eliminated network bursting, indicating the presence of GABAergic neurotransmission. To identify the role of NaV1.1 on neuronal and network activity, cultures were treated with a dose-response of the NaV1.1 potentiator δ-theraphotoxin-Hm1a. This resulted in a strong increase in firing rates of putative GABAergic neurons, an increase in the intraburst firing rate, and eliminated network bursting. These results validate that potentiation of NaV1.1 in Dravet patient iPSC-derived neurons results in decreased firing synchrony in neuronal networks through increased GABAergic neuron activity and support the use of human neurons and HD-MEAs as viable high-throughput electrophysiological platform to enable therapeutic discovery.}, keywords = {}, pubstate = {published}, tppubtype = {article} } Dravet syndrome is a developmental and epileptic encephalopathy characterized by seizures, behavioral abnormalities, developmental deficits, and elevated risk of sudden unexpected death in epilepsy (SUDEP). Most patient cases are caused by de novo loss-of-function mutations in the gene SCN1A, causing a haploinsufficiency of the alpha subunit of the voltage-gated sodium channel NaV1.1. Within the brain, NaV1.1 is primarily localized to the axons of inhibitory neurons, and decreased NaV1.1 function is hypothesized to reduce GABAergic inhibitory neurotransmission within the brain, driving neuronal network hyperexcitability and subsequent pathology. We have developed a human in vitro model of Dravet syndrome using differentiated neurons derived from patient iPSC and enriched for GABA expressing neurons. Neurons were plated on high definition multielectrode arrays (HD-MEAs), permitting recordings from the same cultures over the 7-weeks duration of study at the network, single cell, and subcellular resolution. Using this capability, we characterized the features of axonal morphology and physiology. Neurons developed increased spiking activity and synchronous network bursting. Recordings were processed through a spike sorting pipeline for curation of single unit activity and to assess the effects of pharmacological treatments. At 7-weeks, the application of the GABAAR receptor agonist muscimol eliminated network bursting, indicating the presence of GABAergic neurotransmission. To identify the role of NaV1.1 on neuronal and network activity, cultures were treated with a dose-response of the NaV1.1 potentiator δ-theraphotoxin-Hm1a. This resulted in a strong increase in firing rates of putative GABAergic neurons, an increase in the intraburst firing rate, and eliminated network bursting. These results validate that potentiation of NaV1.1 in Dravet patient iPSC-derived neurons results in decreased firing synchrony in neuronal networks through increased GABAergic neuron activity and support the use of human neurons and HD-MEAs as viable high-throughput electrophysiological platform to enable therapeutic discovery. |
| 6. | Silverman, Jill L; Fenton, Timothy; Haouchine, Olivia; Hallam, Elizabeth; Smith, Emily; Jackson, Kiya; Rahbarian, Darlene; Canales, Cesar; Adhikari, Anna; Nord, Alex; Ben-Shalom, Roy: Hyperexcitability and translational phenotypes in a preclinical model of SYNGAP1mutations. In: Research Square, 2023. (Type: Journal Article | Abstract | Links | BibTeX) @article{Silverman2023, title = {Hyperexcitability and translational phenotypes in a preclinical model of SYNGAP1mutations}, author = {Jill L. Silverman and Timothy Fenton and Olivia Haouchine and Elizabeth Hallam and Emily Smith and Kiya Jackson and Darlene Rahbarian and Cesar Canales and Anna Adhikari and Alex Nord and Roy Ben-Shalom}, url = {https://www.researchsquare.com/article/rs-3246655/v1}, doi = {https://doi.org/10.21203/rs.3.rs-3246655/v1}, year = {2023}, date = {2023-09-13}, journal = {Research Square}, abstract = {SYNGAP1is a critical gene for neuronal development, synaptic structure, and function. Although rare, the disruption of SYNGAP1directly causes a genetically identi able neurodevelopmental disorder (NDD) called SYNGAP1-related intellectual disability. Without functional SynGAP1 protein, patients present with intellectual disability, motor impairments, and epilepsy. Previous work using mouse models with a variety of germline and conditional mutations has helped delineate SynGAP1’s critical roles in neuronal structure and function, as well as key biochemical signaling pathways essential to synapse integrity. Homozygous loss of SYNGAP1is embryonically lethal. Heterozygous mutations of SynGAP1result in a broad range of phenotypes including increased locomotor activity, impaired working spatial memory, impaired cued fear memory, and increased stereotypic behavior. Ourinvivofunctional data, using the original germline mutation mouse line from the Huganir laboratory, corroborated robust hyperactivity and learning and memory de cits. Here, we describe impairments in the translational biomarker domain of sleep, characterized using neurophysiological data collected with wireless telemetric electroencephalography (EEG). We discoveredSyngap1+/− mice exhibited elevated spike trains in both number and duration, in addition to elevated power, most notably in the delta power band. Primary neurons fromSyngap1+/− mice displayed increased network ring activity, greater spikes per burst, and shorter inter-burst intervals between peaks using high density micro-electrode arrays (HD-MEA). This work is translational, innovative, and highly signi cant as it outlines functional impairments in Syngap1mutant mice. Simultaneously, the work utilized untethered, wireless neurophysiology that can discover potential biomarkers of Syngap1RID, for clinical trials, as it has done with other NDDs. Our work is substantial forward progress toward translational work for SynGAP1R-ID as it bridges in-vitroelectrophysiological neuronal activity and function with invivoneurophysiological brain activity and function. These data elucidate multiple quantitative, translational biomarkers invivoand invitrofor the development of treatments for SYNGAP1-related intellectual disability.}, keywords = {}, pubstate = {published}, tppubtype = {article} } SYNGAP1is a critical gene for neuronal development, synaptic structure, and function. Although rare, the disruption of SYNGAP1directly causes a genetically identi able neurodevelopmental disorder (NDD) called SYNGAP1-related intellectual disability. Without functional SynGAP1 protein, patients present with intellectual disability, motor impairments, and epilepsy. Previous work using mouse models with a variety of germline and conditional mutations has helped delineate SynGAP1’s critical roles in neuronal structure and function, as well as key biochemical signaling pathways essential to synapse integrity. Homozygous loss of SYNGAP1is embryonically lethal. Heterozygous mutations of SynGAP1result in a broad range of phenotypes including increased locomotor activity, impaired working spatial memory, impaired cued fear memory, and increased stereotypic behavior. Ourinvivofunctional data, using the original germline mutation mouse line from the Huganir laboratory, corroborated robust hyperactivity and learning and memory de cits. Here, we describe impairments in the translational biomarker domain of sleep, characterized using neurophysiological data collected with wireless telemetric electroencephalography (EEG). We discoveredSyngap1+/− mice exhibited elevated spike trains in both number and duration, in addition to elevated power, most notably in the delta power band. Primary neurons fromSyngap1+/− mice displayed increased network ring activity, greater spikes per burst, and shorter inter-burst intervals between peaks using high density micro-electrode arrays (HD-MEA). This work is translational, innovative, and highly signi cant as it outlines functional impairments in Syngap1mutant mice. Simultaneously, the work utilized untethered, wireless neurophysiology that can discover potential biomarkers of Syngap1RID, for clinical trials, as it has done with other NDDs. Our work is substantial forward progress toward translational work for SynGAP1R-ID as it bridges in-vitroelectrophysiological neuronal activity and function with invivoneurophysiological brain activity and function. These data elucidate multiple quantitative, translational biomarkers invivoand invitrofor the development of treatments for SYNGAP1-related intellectual disability. |
| 7. | Xu, He Jax; Yao, Yao; Yao, Fenyong; Chen, Jiehui; Li, Meishi; Yang, Xianfa; Li, Sheng; Lu, Fangru; Hu, Ping; He, Shuijin; Peng, Guangdun; Jing, Naihe: Generation of functional posterior spinal motor neurons from hPSCs-derived human spinal cord neural progenitor cells. In: Cell Regeneration, 2023. (Type: Journal Article | Abstract | Links | BibTeX) @article{Xu2023, title = {Generation of functional posterior spinal motor neurons from hPSCs-derived human spinal cord neural progenitor cells}, author = {He Jax Xu and Yao Yao and Fenyong Yao and Jiehui Chen and Meishi Li and Xianfa Yang and Sheng Li and Fangru Lu and Ping Hu and Shuijin He and Guangdun Peng and Naihe Jing}, url = {https://cellregeneration.springeropen.com/articles/10.1186/s13619-023-00159-6}, doi = {10.1186/s13619-023-00159-6}, year = {2023}, date = {2023-03-23}, journal = {Cell Regeneration}, abstract = {Spinal motor neurons deficiency results in a series of devastating disorders such as amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA) and spinal cord injury (SCI). These disorders are currently incurable, while human pluripotent stem cells (hPSCs)-derived spinal motor neurons are promising but suffered from inappropriate regional identity and functional immaturity for the study and treatment of posterior spinal cord related injuries. In this study, we have established human spinal cord neural progenitor cells (hSCNPCs) via hPSCs differentiated neuromesodermal progenitors (NMPs) and demonstrated the hSCNPCs can be continuously expanded up to 40 passages. hSCNPCs can be rapidly differentiated into posterior spinal motor neurons with high efficiency. The functional maturity has been examined in detail. Moreover, a co-culture scheme which is compatible for both neural and muscular differentiation is developed to mimic the neuromuscular junction (NMJ) formation in vitro. Together, these studies highlight the potential avenues for generating clinically relevant spinal motor neurons and modeling neuromuscular diseases through our defined hSCNPCs.}, keywords = {}, pubstate = {published}, tppubtype = {article} } Spinal motor neurons deficiency results in a series of devastating disorders such as amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA) and spinal cord injury (SCI). These disorders are currently incurable, while human pluripotent stem cells (hPSCs)-derived spinal motor neurons are promising but suffered from inappropriate regional identity and functional immaturity for the study and treatment of posterior spinal cord related injuries. In this study, we have established human spinal cord neural progenitor cells (hSCNPCs) via hPSCs differentiated neuromesodermal progenitors (NMPs) and demonstrated the hSCNPCs can be continuously expanded up to 40 passages. hSCNPCs can be rapidly differentiated into posterior spinal motor neurons with high efficiency. The functional maturity has been examined in detail. Moreover, a co-culture scheme which is compatible for both neural and muscular differentiation is developed to mimic the neuromuscular junction (NMJ) formation in vitro. Together, these studies highlight the potential avenues for generating clinically relevant spinal motor neurons and modeling neuromuscular diseases through our defined hSCNPCs. |
| 8. | Lin, Waka; Shiomoto, Shusaku; Yamada, Saki; Watanabe, Hikaru; Kawashima, Yudai; Eguchi, Yuichi; Muramatsu, Koichi; Sekino, Yuko: Dendritic spine formation and synapse maturation in transcription factor-induced human iPSC-derived neurons. In: iScience, 2023. (Type: Journal Article | Abstract | Links | BibTeX) @article{Lin2023, title = {Dendritic spine formation and synapse maturation in transcription factor-induced human iPSC-derived neurons}, author = {Waka Lin and Shusaku Shiomoto and Saki Yamada and Hikaru Watanabe and Yudai Kawashima and Yuichi Eguchi and Koichi Muramatsu and Yuko Sekino}, url = {https://pubmed.ncbi.nlm.nih.gov/37034988/}, year = {2023}, date = {2023-02-27}, journal = {iScience}, abstract = {Synaptic maturation is reportedly limited in human induced pluripotent stem cell (iPSC)-derived neurons. Notably, their ability to reach postnatal-like stages and form dendritic spines has been difficult to demonstrate unless using long-term cultured organoids. Recent transcription factor (TF)-based induction methods allow the accelerated generation of differentiated neurons, which offers an unprecedented opportunity to address further progression into late developmental stages. Herein, we report on a comprehensive time-course study of TF-induced iPSC neurons cultured in vitro through an intrinsic maturation program following neurogenesis. Moreover, we determined the transcriptional and morphological sequences of key developmental events associated with spinogenesis, including the conversion of drebrin to its brain-specific isoform A and the N-methyl-D-aspartate (NMDA) receptor subunit switch. TF-induced iPSC neurons successfully acquired structural and functional synaptic maturity, which will critically expand their utility in modeling higher brain functions and disorders.}, keywords = {}, pubstate = {published}, tppubtype = {article} } Synaptic maturation is reportedly limited in human induced pluripotent stem cell (iPSC)-derived neurons. Notably, their ability to reach postnatal-like stages and form dendritic spines has been difficult to demonstrate unless using long-term cultured organoids. Recent transcription factor (TF)-based induction methods allow the accelerated generation of differentiated neurons, which offers an unprecedented opportunity to address further progression into late developmental stages. Herein, we report on a comprehensive time-course study of TF-induced iPSC neurons cultured in vitro through an intrinsic maturation program following neurogenesis. Moreover, we determined the transcriptional and morphological sequences of key developmental events associated with spinogenesis, including the conversion of drebrin to its brain-specific isoform A and the N-methyl-D-aspartate (NMDA) receptor subunit switch. TF-induced iPSC neurons successfully acquired structural and functional synaptic maturity, which will critically expand their utility in modeling higher brain functions and disorders. |
| 9. | Lent, Jonas Van; Vendredy, Leen; Adriaenssens, Elias; Authier, Tatiana Da Silva; Asselbergh, Bob; Kaji, Marcus; Weckhuysen, Sarah; Bosch, Ludo Van Den; Baets, Jonathan; Timmerman, Vincent: Downregulation of PMP22 ameliorates myelin defects in iPSC-derived human organoid cultures of CMT1A. In: Brain, 2022. (Type: Journal Article | Abstract | Links | BibTeX) @article{VanLent2022, title = {Downregulation of PMP22 ameliorates myelin defects in iPSC-derived human organoid cultures of CMT1A}, author = {Jonas Van Lent and Leen Vendredy and Elias Adriaenssens and Tatiana Da Silva Authier and Bob Asselbergh and Marcus Kaji and Sarah Weckhuysen and Ludo Van Den Bosch and Jonathan Baets and Vincent Timmerman}, url = {https://academic.oup.com/brain/advance-article/doi/10.1093/brain/awac475/6895197?login=false}, doi = {https://doi.org/10.1093/brain/awac475}, year = {2022}, date = {2022-12-12}, journal = {Brain}, abstract = {Charcot-Marie-Tooth (CMT) disease is the most common inherited disorder of the peripheral nervous system. CMT1A accounts for 40-50% of all cases and is caused by a duplication of the PMP22 gene on chromosome 17, leading to dysmyelination in the peripheral nervous system. Patient-derived models to study such myelination defects are lacking as the in vitro generation of human myelinating Schwann cells has proven to be particularly challenging. Here, we present an iPSC-derived organoid culture, containing various cell types of the peripheral nervous system, including myelinating human Schwann cells, which mimics the human peripheral nervous system. Single-cell analysis confirmed the peripheral nervous system-like cellular composition and provides insight into the developmental trajectory. We used this organoid-model to study disease signatures of CMT1A, revealing early ultrastructural myelin alterations, including increased myelin periodic line distance and hypermyelination of small axons. Furthermore, we observed the presence of onion bulb-like formations in a later developmental stage. These hallmarks were not present in the for CMT1A-corrected isogenic line or in a CMT2A iPSC line, supporting the notion that these alterations are specific to CMT1A. Downregulation of PMP22 expression using short-hairpin RNAs or a combinatorial drug consisting of baclofen, naltrexone hydrochloride and D-sorbitol, was able to ameliorate the myelin defects in CMT1A-organoids. In summary, this self-organizing organoid model is able to capture biologically meaningful features of the disease and capture the physiological complexity, forms an excellent model to study demyelinating diseases, and supports the therapeutic approach of reducing PMP22 expression.}, keywords = {}, pubstate = {published}, tppubtype = {article} } Charcot-Marie-Tooth (CMT) disease is the most common inherited disorder of the peripheral nervous system. CMT1A accounts for 40-50% of all cases and is caused by a duplication of the PMP22 gene on chromosome 17, leading to dysmyelination in the peripheral nervous system. Patient-derived models to study such myelination defects are lacking as the in vitro generation of human myelinating Schwann cells has proven to be particularly challenging. Here, we present an iPSC-derived organoid culture, containing various cell types of the peripheral nervous system, including myelinating human Schwann cells, which mimics the human peripheral nervous system. Single-cell analysis confirmed the peripheral nervous system-like cellular composition and provides insight into the developmental trajectory. We used this organoid-model to study disease signatures of CMT1A, revealing early ultrastructural myelin alterations, including increased myelin periodic line distance and hypermyelination of small axons. Furthermore, we observed the presence of onion bulb-like formations in a later developmental stage. These hallmarks were not present in the for CMT1A-corrected isogenic line or in a CMT2A iPSC line, supporting the notion that these alterations are specific to CMT1A. Downregulation of PMP22 expression using short-hairpin RNAs or a combinatorial drug consisting of baclofen, naltrexone hydrochloride and D-sorbitol, was able to ameliorate the myelin defects in CMT1A-organoids. In summary, this self-organizing organoid model is able to capture biologically meaningful features of the disease and capture the physiological complexity, forms an excellent model to study demyelinating diseases, and supports the therapeutic approach of reducing PMP22 expression. |
| 10. | Buccino Alessio Paolo; Damart, Tanguy; Bartram Julian; Mandge Darshan; Xue Xiaohan; Zbili Mickael; Gänswein Tobias; Jaquier Aurélien; Emmenegger Vishalini; Markram Henry; Hierlemann Andreas; Van Geit Werner. : A multi-modal fitting approach to construct single-neuron models with patch clamp and high-density microelectrode arrays. In: bioRxiv, 2022. (Type: Journal Article | Abstract | Links | BibTeX) @article{Buccino2022, title = {A multi-modal fitting approach to construct single-neuron models with patch clamp and high-density microelectrode arrays}, author = {Buccino, Alessio Paolo; Damart, Tanguy; Bartram, Julian; Mandge, Darshan; Xue, Xiaohan; Zbili, Mickael; Gänswein, Tobias; Jaquier, Aurélien; Emmenegger, Vishalini; Markram, Henry; Hierlemann, Andreas; Van Geit, Werner.}, doi = {https://doi.org/10.1101/2022.08.03.502468}, year = {2022}, date = {2022-08-11}, journal = {bioRxiv}, abstract = {In computational neuroscience, multicompartment models are among the most biophysically realistic representations of single neurons. Constructing such models usually involves the use of the patch-clamp technique to record somatic voltage signals under different experimental conditions. The experimental data are then used to fit the many parameters of the model. While patching of the soma is currently the gold-standard approach to build multicompartment models, several studies have also evidenced a richness of dynamics in dendritic and axonal sections. Recording from the soma alone makes it hard to observe and correctly parameterize the activity of non-somatic compartments. In order to provide a richer set of data as input to multicompartment models, we here investigate the combination of somatic patch-clamp recordings with recordings of high-density micro-electrode arrays (HD-MEAs). HD-MEAs enable the observation of extracellular potentials and neural activity of neuronal compartments at sub-cellular resolution. In this work, we introduce a novel framework to combine patch-clamp and HD-MEA data to construct multicompartment models. We first validate our method on a ground-truth model with known parameters and show that the use of features extracted from extracellular signals, in addition to intracellular ones, yields models enabling better fits than using intracellular features alone. We also demonstrate our procedure using experimental data by constructing cell models from in vitro cell cultures. The proposed multi-modal fitting procedure has the potential to augment the modeling efforts of the computational neuroscience community and to provide the field with neuronal models that are more realistic and can be better validated. Author Summary Multicompartment models are one of the most biophysically detailed representations of single neurons. The vast majority of these models are built using experimental data from somatic recordings. However, neurons are much more than just their soma and one needs recordings from distal neurites to build an accurate model. In this article, we combine the patch-clamp technique with extracellular high-density microelectrode arrays (HD-MEAs) to compensate this shortcoming. In fact, HD-MEAs readouts allow one to record the neuronal signal in the entire axonal arbor. We show that the proposed multi-modal strategy is superior to the use of patch clamp alone using an existing model as ground-truth. Finally, we show an application of this strategy on experimental data from cultured neurons.}, keywords = {}, pubstate = {published}, tppubtype = {article} } In computational neuroscience, multicompartment models are among the most biophysically realistic representations of single neurons. Constructing such models usually involves the use of the patch-clamp technique to record somatic voltage signals under different experimental conditions. The experimental data are then used to fit the many parameters of the model. While patching of the soma is currently the gold-standard approach to build multicompartment models, several studies have also evidenced a richness of dynamics in dendritic and axonal sections. Recording from the soma alone makes it hard to observe and correctly parameterize the activity of non-somatic compartments. In order to provide a richer set of data as input to multicompartment models, we here investigate the combination of somatic patch-clamp recordings with recordings of high-density micro-electrode arrays (HD-MEAs). HD-MEAs enable the observation of extracellular potentials and neural activity of neuronal compartments at sub-cellular resolution. In this work, we introduce a novel framework to combine patch-clamp and HD-MEA data to construct multicompartment models. We first validate our method on a ground-truth model with known parameters and show that the use of features extracted from extracellular signals, in addition to intracellular ones, yields models enabling better fits than using intracellular features alone. We also demonstrate our procedure using experimental data by constructing cell models from in vitro cell cultures. The proposed multi-modal fitting procedure has the potential to augment the modeling efforts of the computational neuroscience community and to provide the field with neuronal models that are more realistic and can be better validated. Author Summary Multicompartment models are one of the most biophysically detailed representations of single neurons. The vast majority of these models are built using experimental data from somatic recordings. However, neurons are much more than just their soma and one needs recordings from distal neurites to build an accurate model. In this article, we combine the patch-clamp technique with extracellular high-density microelectrode arrays (HD-MEAs) to compensate this shortcoming. In fact, HD-MEAs readouts allow one to record the neuronal signal in the entire axonal arbor. We show that the proposed multi-modal strategy is superior to the use of patch clamp alone using an existing model as ground-truth. Finally, we show an application of this strategy on experimental data from cultured neurons. |
| 11. | Ricci, Chiara; Frey, Urs; Obien, Marie Engelene J: MAPSYNE: Miniaturized micropipette system combined with high-density microelectrode arrays for automated manipulation of neuronal networks in-vitro. In: IEEE, 2020. (Type: Journal Article | Abstract | Links | BibTeX) @article{Ricci2020, title = {MAPSYNE: Miniaturized micropipette system combined with high-density microelectrode arrays for automated manipulation of neuronal networks in-vitro}, author = {Chiara Ricci and Urs Frey and Marie Engelene J. Obien}, url = {https://ieeexplore.ieee.org/document/9175797/}, doi = {10.1109/EMBC44109.2020.9175797}, year = {2020}, date = {2020-10-08}, journal = {IEEE}, abstract = {We present MAPSYNE, a miniaturized and automated system combining a high-density microelectrode array (HD-MEA) and a movable micropipette for studying, monitoring, and perturbing neurons in vitro. The system involves an all-electrical approach to automatically move a glass micropipette towards a target location on the HD-MEA surface, without the need for a microscope. Two methods of performing blind navigation are employed, (i) stop-measure-go approach wherein the pipette moves for a predefined distance before measuring its location then the process is repeated until the pipette reaches its destination, and (ii) predictive approach wherein the pipette is continuously tracked and moved. This automated system can be applied for unsupervised single-cell manipulation of neurons in a network, such as electroporation and local delivery of compounds.}, keywords = {}, pubstate = {published}, tppubtype = {article} } We present MAPSYNE, a miniaturized and automated system combining a high-density microelectrode array (HD-MEA) and a movable micropipette for studying, monitoring, and perturbing neurons in vitro. The system involves an all-electrical approach to automatically move a glass micropipette towards a target location on the HD-MEA surface, without the need for a microscope. Two methods of performing blind navigation are employed, (i) stop-measure-go approach wherein the pipette moves for a predefined distance before measuring its location then the process is repeated until the pipette reaches its destination, and (ii) predictive approach wherein the pipette is continuously tracked and moved. This automated system can be applied for unsupervised single-cell manipulation of neurons in a network, such as electroporation and local delivery of compounds. |
Hruska-Plochan, Marian; Wiersma, Vera I; Betz, Katharina M; Mallona, Izaskun; Ronchi, Silvia; Maniecka, Zuzanna; Hock, Eva-Maria; Tantardini, Elena; Laferriere, Florent; Sahadevan, Sonu; Hoop, Vanessa; Delvendahl, Igor; Pérez-Berlanga, Manuela; Gatta, Beatrice; Panatta, Martina; van der Bourg, Alexander; Bohaciakova, Dasa; Sharma, Puneet; Vos, Laura De; Frontzek, Karl; Aguzzi, Adriano; Lashley, Tammaryn; Robinson, Mark D; Karayannis, Theofanis; Mueller, Martin; Hierlemann, Andreas; Polymenidou, Magdalini A model of human neural networks reveals NPTX2 pathology in ALS and FTLD Journal Article Nature, 2024. Abstract | Links | BibTeX | タグ: Activity Scan Assay, HD-MEA, IPSC, MaxOne, MEA Technology, Network Assay, Primary Neuronal Cell Culture, Spike Sorting Ryu, Seungmi; Weber, Claire; Chu, Pei-Hsuan; Ernest, Ben; Jovanovic, Vukasin M; Deng, Tao; Slamecka, Jaroslav; Hong, Hyenjong; Jethmalani, Yogita; Baskir, Hannah M; Inman, Jason; Braisted, John; Hirst, Marissa B; Simeonov, Anton; Voss, Ty C; Tristan, Carlos A; Singeç, Ilyas Stress-free cell aggregation by using the CEPT cocktail enhances embryoid body and organoid fitness Journal Article Biofabrication, 2023. Abstract | Links | BibTeX | タグ: 3D Culture, Activity Scan Assay, HD-MEA, MEA Technology, Organoids Elliott, Matthew A T; Schweiger, Hunter E; Robbins, Ash; Vera-Choqqueccota, Samira; Ehrlich, Drew; Hernandez, Sebastian; Voitiuk, Kateryna; Geng, Jinghui; Sevetson, Jess L; Core, Cordero; Rosen, Yohei M; Teodorescu, Mircea; Wagner, Nico O; Haussler, David; Mostajo-Radji, Mohammed A Internet-Connected Cortical Organoids for Project-Based Stem Cell and Neuroscience Education Journal Article eNeuro, 2023. Abstract | Links | BibTeX | タグ: 3D Culture, Activity Scan Assay, HD-MEA, IPSC, MaxOne, MEA Technology, Organoids, Spike Sorting, Stimulation Sawada, Tomoyo; Barbosa, André R; Araujo, Bruno; McCord, Alejandra E; D’Ignazio, Laura; Benjamin, Kynon J M; Sheehan, Bonna; Zabolocki, Michael; Feltrin, Arthur; Arora, Ria; Brandtjen, Anna C; Kleinman, Joel E; Hyde, Thomas M; Bardy, Cedric; Weinberger, Daniel R; Paquola, Apuã C M; Erwin, Jennifer A American Journal of Psychiatry, 2023, ISSN: 0002-953X. Abstract | Links | BibTeX | タグ: 3D Culture, Activity Scan Assay, HD-MEA, MaxTwo, Network Assay, Organoids Kelley, Matt R; Chipman, Laura B; Asano, Shoh; Knott, Matthew; Howard, Samantha T; Berg, Allison P bioRxiv, 2023. Abstract | Links | BibTeX | タグ: Activity Scan Assay, Axon Tracking Assay, HD-MEA, IPSC, MaxTwo, MEA Technology, Network Assay, Spike Sorting Silverman, Jill L; Fenton, Timothy; Haouchine, Olivia; Hallam, Elizabeth; Smith, Emily; Jackson, Kiya; Rahbarian, Darlene; Canales, Cesar; Adhikari, Anna; Nord, Alex; Ben-Shalom, Roy Hyperexcitability and translational phenotypes in a preclinical model of SYNGAP1mutations Journal Article Research Square, 2023. Abstract | Links | BibTeX | タグ: 2D Neuronal Culture, Activity Scan Assay, HD-MEA, MaxOne, Network Assay, Primary Neuronal Cell Culture Xu, He Jax; Yao, Yao; Yao, Fenyong; Chen, Jiehui; Li, Meishi; Yang, Xianfa; Li, Sheng; Lu, Fangru; Hu, Ping; He, Shuijin; Peng, Guangdun; Jing, Naihe Generation of functional posterior spinal motor neurons from hPSCs-derived human spinal cord neural progenitor cells Journal Article Cell Regeneration, 2023. Abstract | Links | BibTeX | タグ: 2D Neuronal Culture, Activity Scan Assay, Axon Tracking Assay, HD-MEA, IPSC, MaxOne, MEA Technology, Network Assay, Organoids Lin, Waka; Shiomoto, Shusaku; Yamada, Saki; Watanabe, Hikaru; Kawashima, Yudai; Eguchi, Yuichi; Muramatsu, Koichi; Sekino, Yuko Dendritic spine formation and synapse maturation in transcription factor-induced human iPSC-derived neurons Journal Article iScience, 2023. Abstract | Links | BibTeX | タグ: Activity Scan Assay, MaxTwo, Network Assay Lent, Jonas Van; Vendredy, Leen; Adriaenssens, Elias; Authier, Tatiana Da Silva; Asselbergh, Bob; Kaji, Marcus; Weckhuysen, Sarah; Bosch, Ludo Van Den; Baets, Jonathan; Timmerman, Vincent Downregulation of PMP22 ameliorates myelin defects in iPSC-derived human organoid cultures of CMT1A Journal Article Brain, 2022. Abstract | Links | BibTeX | タグ: Activity Scan Assay, Axon Tracking Assay, MaxTwo, Network Assay, Organoids Buccino Alessio Paolo; Damart, Tanguy; Bartram Julian; Mandge Darshan; Xue Xiaohan; Zbili Mickael; Gänswein Tobias; Jaquier Aurélien; Emmenegger Vishalini; Markram Henry; Hierlemann Andreas; Van Geit Werner. A multi-modal fitting approach to construct single-neuron models with patch clamp and high-density microelectrode arrays Journal Article bioRxiv, 2022. Abstract | Links | BibTeX | タグ: 2D Neuronal Culture, Activity Scan Assay, Axon Tracking Assay, HD-MEA, MaxOne, Other Tissues, Publication, Stimulation Assay Ricci, Chiara; Frey, Urs; Obien, Marie Engelene J IEEE, 2020. Abstract | Links | BibTeX | タグ: Activity Scan Assay, HD-MEA, MaxOne, MEA Technology
2024
![]()
title = {A model of human neural networks reveals NPTX2 pathology in ALS and FTLD},
author = {Marian Hruska-Plochan and Vera I. Wiersma and Katharina M. Betz and Izaskun Mallona and Silvia Ronchi and Zuzanna Maniecka and Eva-Maria Hock and Elena Tantardini and Florent Laferriere and Sonu Sahadevan and Vanessa Hoop and Igor Delvendahl and Manuela Pérez-Berlanga and Beatrice Gatta and Martina Panatta and Alexander van der Bourg and Dasa Bohaciakova and Puneet Sharma and Laura De Vos and Karl Frontzek and Adriano Aguzzi and Tammaryn Lashley and Mark D. Robinson and Theofanis Karayannis and Martin Mueller and Andreas Hierlemann and Magdalini Polymenidou },
url = {https://www.nature.com/articles/s41586-024-07042-7},
doi = {10.1038/s41586-024-07042-7},
year = {2024},
date = {2024-02-14},
journal = {Nature},
abstract = {Human cellular models of neurodegeneration require reproducibility and longevity, which is necessary for simulating age-dependent diseases. Such systems are particularly needed for TDP-43 proteinopathies1, which involve human-specific mechanisms that cannot be directly studied in animal models. Here, to explore the emergence and consequences of TDP-43 pathologies, we generated induced pluripotent stem cell-derived, colony morphology neural stem cells (iCoMoNSCs) via manual selection of neural precursors. Single-cell transcriptomics and comparison to independent neural stem cells showed that iCoMoNSCs are uniquely homogenous and self-renewing. Differentiated iCoMoNSCs formed a self-organized multicellular system consisting of synaptically connected and electrophysiologically active neurons, which matured into long-lived functional networks (which we designate iNets). Neuronal and glial maturation in iNets was similar to that of cortical organoids. Overexpression of wild-type TDP-43 in a minority of neurons within iNets led to progressive fragmentation and aggregation of the protein, resulting in a partial loss of function and neurotoxicity. Single-cell transcriptomics revealed a novel set of misregulated RNA targets in TDP-43-overexpressing neurons and in patients with TDP-43 proteinopathies exhibiting a loss of nuclear TDP-43. The strongest misregulated target encoded the synaptic protein NPTX2, the levels of which are controlled by TDP-43 binding on its 3′ untranslated region. When NPTX2 was overexpressed in iNets, it exhibited neurotoxicity, whereas correcting NPTX2 misregulation partially rescued neurons from TDP-43-induced neurodegeneration. Notably, NPTX2 was consistently misaccumulated in neurons from patients with amyotrophic lateral sclerosis and frontotemporal lobar degeneration with TDP-43 pathology. Our work directly links TDP-43 misregulation and NPTX2 accumulation, thereby revealing a TDP-43-dependent pathway of neurotoxicity.},
keywords = {Activity Scan Assay, HD-MEA, IPSC, MaxOne, MEA Technology, Network Assay, Primary Neuronal Cell Culture, Spike Sorting},
pubstate = {published},
tppubtype = {article}
}
2023

title = {Stress-free cell aggregation by using the CEPT cocktail enhances embryoid body and organoid fitness},
author = {Seungmi Ryu and Claire Weber and Pei-Hsuan Chu and Ben Ernest and Vukasin M Jovanovic and Tao Deng and Jaroslav Slamecka and Hyenjong Hong and Yogita Jethmalani and Hannah M Baskir and Jason Inman and John Braisted and Marissa B Hirst and Anton Simeonov and Ty C Voss and Carlos A Tristan and Ilyas Singeç},
url = {https://dx.doi.org/10.1088/1758-5090/ad0d13},
doi = {10.1088/1758-5090/ad0d13},
year = {2023},
date = {2023-12-11},
journal = {Biofabrication},
abstract = {Embryoid bodies (EBs) and self-organizing organoids derived from human pluripotent stem cells (hPSCs) recapitulate tissue development in a dish and hold great promise for disease modeling and drug development. However, current protocols are hampered by cellular stress and apoptosis during cell aggregation, resulting in variability and impaired cell differentiation. Here, we demonstrate that EBs and various organoid models (e.g., brain, gut, kidney) can be optimized by using the small molecule cocktail named CEPT (chroman 1, emricasan, polyamines, trans-ISRIB), a polypharmacological approach that ensures cytoprotection and cell survival. Application of CEPT for just 24 h during cell aggregation has long-lasting consequences affecting morphogenesis, gene expression, cellular differentiation, and organoid function. Various qualification methods confirmed that CEPT treatment enhanced experimental reproducibility and consistently improved EB and organoid fitness as compared to the widely used ROCK inhibitor Y-27632. Collectively, we discovered that stress-free cell aggregation and superior cell survival in the presence of CEPT are critical quality control determinants that establish a robust foundation for bioengineering complex tissue and organ models.},
keywords = {3D Culture, Activity Scan Assay, HD-MEA, MEA Technology, Organoids},
pubstate = {published},
tppubtype = {article}
}
![]()
title = {Internet-Connected Cortical Organoids for Project-Based Stem Cell and Neuroscience Education},
author = {Matthew A. T. Elliott and Hunter E. Schweiger and Ash Robbins and Samira Vera-Choqqueccota and Drew Ehrlich and Sebastian Hernandez and Kateryna Voitiuk and Jinghui Geng and Jess L. Sevetson and Cordero Core and Yohei M. Rosen and Mircea Teodorescu and Nico O. Wagner and David Haussler and Mohammed A. Mostajo-Radji},
url = {https://www.eneuro.org/lookup/doi/10.1523/ENEURO.0308-23.2023},
doi = {10.1523/ENEURO.0308-23.2023},
year = {2023},
date = {2023-11-28},
journal = {eNeuro},
abstract = {The introduction of Internet-connected technologies to the classroom has the potential to revolutionize STEM education by allowing students to perform experiments in complex models that are unattainable in traditional teaching laboratories. By connecting laboratory equipment to the cloud, we introduce students to experimentation in pluripotent stem cell (PSC)-derived cortical organoids in two different settings: using microscopy to monitor organoid growth in an introductory tissue culture course and using high-density (HD) multielectrode arrays (MEAs) to perform neuronal stimulation and recording in an advanced neuroscience mathematics course. We demonstrate that this approach develops interest in stem cell and neuroscience in the students of both courses. All together, we propose cloud technologies as an effective and scalable approach for complex project-based university training.},
keywords = {3D Culture, Activity Scan Assay, HD-MEA, IPSC, MaxOne, MEA Technology, Organoids, Spike Sorting, Stimulation},
pubstate = {published},
tppubtype = {article}
}

title = {Recapitulation of Perturbed Striatal Gene Expression Dynamics of Donor’s Brains With Ventral Forebrain Organoids Derived From the Same Individuals With Schizophrenia},
author = {Tomoyo Sawada and André R. Barbosa and Bruno Araujo and Alejandra E. McCord and Laura D’Ignazio and Kynon J.M. Benjamin and Bonna Sheehan and Michael Zabolocki and Arthur Feltrin and Ria Arora and Anna C. Brandtjen and Joel E. Kleinman and Thomas M. Hyde and Cedric Bardy and Daniel R. Weinberger and Apuã C.M. Paquola and Jennifer A. Erwin},
url = {https://ajp.psychiatryonline.org/doi/10.1176/appi.ajp.20220723},
doi = {10.1176/appi.ajp.20220723},
issn = {0002-953X},
year = {2023},
date = {2023-11-02},
journal = {American Journal of Psychiatry},
abstract = {Objective:
Schizophrenia is a brain disorder that originates during neurodevelopment and has complex genetic and environmental etiologies. Despite decades of clinical evidence of altered striatal function in affected patients, studies examining its cellular and molecular mechanisms in humans are limited. To explore neurodevelopmental alterations in the striatum associated with schizophrenia, the authors established a method for the differentiation of induced pluripotent stem cells (iPSCs) into ventral forebrain organoids (VFOs).
Methods:
VFOs were generated from postmortem dural fibroblast–derived iPSCs of four individuals with schizophrenia and four neurotypical control individuals for whom postmortem caudate genotypes and transcriptomic data were profiled in the BrainSeq neurogenomics consortium. Individuals were selected such that the two groups had nonoverlapping schizophrenia polygenic risk scores (PRSs).
Results:
Single-cell RNA sequencing analyses of VFOs revealed differences in developmental trajectory between schizophrenia and control individuals in which inhibitory neuronal cells from the patients exhibited accelerated maturation. Furthermore, upregulated genes in inhibitory neurons in schizophrenia VFOs showed a significant overlap with upregulated genes in postmortem caudate tissue of individuals with schizophrenia compared with control individuals, including the donors of the iPSC cohort.
Conclusions:
The findings suggest that striatal neurons derived from high-PRS individuals with schizophrenia carry abnormalities that originated during early brain development and that the VFO model can recapitulate disease-relevant cell type–specific neurodevelopmental phenotypes in a dish.},
keywords = {3D Culture, Activity Scan Assay, HD-MEA, MaxTwo, Network Assay, Organoids},
pubstate = {published},
tppubtype = {article}
}
Schizophrenia is a brain disorder that originates during neurodevelopment and has complex genetic and environmental etiologies. Despite decades of clinical evidence of altered striatal function in affected patients, studies examining its cellular and molecular mechanisms in humans are limited. To explore neurodevelopmental alterations in the striatum associated with schizophrenia, the authors established a method for the differentiation of induced pluripotent stem cells (iPSCs) into ventral forebrain organoids (VFOs).
Methods:
VFOs were generated from postmortem dural fibroblast–derived iPSCs of four individuals with schizophrenia and four neurotypical control individuals for whom postmortem caudate genotypes and transcriptomic data were profiled in the BrainSeq neurogenomics consortium. Individuals were selected such that the two groups had nonoverlapping schizophrenia polygenic risk scores (PRSs).
Results:
Single-cell RNA sequencing analyses of VFOs revealed differences in developmental trajectory between schizophrenia and control individuals in which inhibitory neuronal cells from the patients exhibited accelerated maturation. Furthermore, upregulated genes in inhibitory neurons in schizophrenia VFOs showed a significant overlap with upregulated genes in postmortem caudate tissue of individuals with schizophrenia compared with control individuals, including the donors of the iPSC cohort.
Conclusions:
The findings suggest that striatal neurons derived from high-PRS individuals with schizophrenia carry abnormalities that originated during early brain development and that the VFO model can recapitulate disease-relevant cell type–specific neurodevelopmental phenotypes in a dish.![]()
title = {Potentiating NaV1.1 in Dravet syndrome patient iPSC-derived GABAergic neurons increases neuronal firing frequency and decreases network synchrony},
author = {Matt R Kelley and Laura B Chipman and Shoh Asano and Matthew Knott and Samantha T Howard and Allison P Berg},
url = {https://www.biorxiv.org/content/10.1101/2023.09.28.559990v1},
doi = {10.1101/2023.09.28.559990},
year = {2023},
date = {2023-09-29},
journal = {bioRxiv},
abstract = {Dravet syndrome is a developmental and epileptic encephalopathy characterized by seizures, behavioral abnormalities, developmental deficits, and elevated risk of sudden unexpected death in epilepsy (SUDEP). Most patient cases are caused by de novo loss-of-function mutations in the gene SCN1A, causing a haploinsufficiency of the alpha subunit of the voltage-gated sodium channel NaV1.1. Within the brain, NaV1.1 is primarily localized to the axons of inhibitory neurons, and decreased NaV1.1 function is hypothesized to reduce GABAergic inhibitory neurotransmission within the brain, driving neuronal network hyperexcitability and subsequent pathology. We have developed a human in vitro model of Dravet syndrome using differentiated neurons derived from patient iPSC and enriched for GABA expressing neurons. Neurons were plated on high definition multielectrode arrays (HD-MEAs), permitting recordings from the same cultures over the 7-weeks duration of study at the network, single cell, and subcellular resolution. Using this capability, we characterized the features of axonal morphology and physiology. Neurons developed increased spiking activity and synchronous network bursting. Recordings were processed through a spike sorting pipeline for curation of single unit activity and to assess the effects of pharmacological treatments. At 7-weeks, the application of the GABAAR receptor agonist muscimol eliminated network bursting, indicating the presence of GABAergic neurotransmission. To identify the role of NaV1.1 on neuronal and network activity, cultures were treated with a dose-response of the NaV1.1 potentiator δ-theraphotoxin-Hm1a. This resulted in a strong increase in firing rates of putative GABAergic neurons, an increase in the intraburst firing rate, and eliminated network bursting. These results validate that potentiation of NaV1.1 in Dravet patient iPSC-derived neurons results in decreased firing synchrony in neuronal networks through increased GABAergic neuron activity and support the use of human neurons and HD-MEAs as viable high-throughput electrophysiological platform to enable therapeutic discovery.},
keywords = {Activity Scan Assay, Axon Tracking Assay, HD-MEA, IPSC, MaxTwo, MEA Technology, Network Assay, Spike Sorting},
pubstate = {published},
tppubtype = {article}
}
![]()
title = {Hyperexcitability and translational phenotypes in a preclinical model of SYNGAP1mutations},
author = {Jill L. Silverman and Timothy Fenton and Olivia Haouchine and Elizabeth Hallam and Emily Smith and Kiya Jackson and Darlene Rahbarian and Cesar Canales and Anna Adhikari and Alex Nord and Roy Ben-Shalom},
url = {https://www.researchsquare.com/article/rs-3246655/v1},
doi = {https://doi.org/10.21203/rs.3.rs-3246655/v1},
year = {2023},
date = {2023-09-13},
journal = {Research Square},
abstract = {SYNGAP1is a critical gene for neuronal development, synaptic structure, and function. Although rare, the disruption of SYNGAP1directly causes a genetically identi able neurodevelopmental disorder (NDD) called SYNGAP1-related intellectual disability. Without functional SynGAP1 protein, patients present with intellectual disability, motor impairments, and epilepsy. Previous work using mouse models with a variety of germline and conditional mutations has helped delineate SynGAP1’s critical roles in neuronal structure and function, as well as key biochemical signaling pathways essential to synapse integrity. Homozygous loss of SYNGAP1is embryonically lethal. Heterozygous mutations of SynGAP1result in a broad range of phenotypes including increased locomotor activity, impaired working spatial memory, impaired cued fear memory, and increased stereotypic behavior. Ourinvivofunctional data, using the original germline mutation mouse line from the Huganir laboratory, corroborated robust hyperactivity and learning and memory de cits. Here, we describe impairments in the translational biomarker domain of sleep, characterized using neurophysiological data collected with wireless telemetric electroencephalography (EEG). We discoveredSyngap1+/− mice exhibited elevated spike trains in both number and duration, in addition to elevated power, most notably in the delta power band. Primary neurons fromSyngap1+/− mice displayed increased network ring activity, greater spikes per burst, and shorter inter-burst intervals between peaks using high density micro-electrode arrays (HD-MEA). This work is translational, innovative, and highly signi cant as it outlines functional impairments in Syngap1mutant mice. Simultaneously, the work utilized untethered, wireless neurophysiology that can discover potential biomarkers of Syngap1RID, for clinical trials, as it has done with other NDDs. Our work is substantial forward progress toward translational work for SynGAP1R-ID as it bridges in-vitroelectrophysiological neuronal activity and function with invivoneurophysiological brain activity and function. These data elucidate multiple quantitative, translational biomarkers invivoand invitrofor the development of treatments for SYNGAP1-related intellectual disability.},
keywords = {2D Neuronal Culture, Activity Scan Assay, HD-MEA, MaxOne, Network Assay, Primary Neuronal Cell Culture},
pubstate = {published},
tppubtype = {article}
}

title = {Generation of functional posterior spinal motor neurons from hPSCs-derived human spinal cord neural progenitor cells},
author = {He Jax Xu and Yao Yao and Fenyong Yao and Jiehui Chen and Meishi Li and Xianfa Yang and Sheng Li and Fangru Lu and Ping Hu and Shuijin He and Guangdun Peng and Naihe Jing},
url = {https://cellregeneration.springeropen.com/articles/10.1186/s13619-023-00159-6},
doi = {10.1186/s13619-023-00159-6},
year = {2023},
date = {2023-03-23},
journal = {Cell Regeneration},
abstract = {Spinal motor neurons deficiency results in a series of devastating disorders such as amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA) and spinal cord injury (SCI). These disorders are currently incurable, while human pluripotent stem cells (hPSCs)-derived spinal motor neurons are promising but suffered from inappropriate regional identity and functional immaturity for the study and treatment of posterior spinal cord related injuries. In this study, we have established human spinal cord neural progenitor cells (hSCNPCs) via hPSCs differentiated neuromesodermal progenitors (NMPs) and demonstrated the hSCNPCs can be continuously expanded up to 40 passages. hSCNPCs can be rapidly differentiated into posterior spinal motor neurons with high efficiency. The functional maturity has been examined in detail. Moreover, a co-culture scheme which is compatible for both neural and muscular differentiation is developed to mimic the neuromuscular junction (NMJ) formation in vitro. Together, these studies highlight the potential avenues for generating clinically relevant spinal motor neurons and modeling neuromuscular diseases through our defined hSCNPCs.},
keywords = {2D Neuronal Culture, Activity Scan Assay, Axon Tracking Assay, HD-MEA, IPSC, MaxOne, MEA Technology, Network Assay, Organoids},
pubstate = {published},
tppubtype = {article}
}

title = {Dendritic spine formation and synapse maturation in transcription factor-induced human iPSC-derived neurons},
author = {Waka Lin and Shusaku Shiomoto and Saki Yamada and Hikaru Watanabe and Yudai Kawashima and Yuichi Eguchi and Koichi Muramatsu and Yuko Sekino},
url = {https://pubmed.ncbi.nlm.nih.gov/37034988/},
year = {2023},
date = {2023-02-27},
journal = {iScience},
abstract = {Synaptic maturation is reportedly limited in human induced pluripotent stem cell (iPSC)-derived neurons. Notably, their ability to reach postnatal-like stages and form dendritic spines has been difficult to demonstrate unless using long-term cultured organoids. Recent transcription factor (TF)-based induction methods allow the accelerated generation of differentiated neurons, which offers an unprecedented opportunity to address further progression into late developmental stages. Herein, we report on a comprehensive time-course study of TF-induced iPSC neurons cultured in vitro through an intrinsic maturation program following neurogenesis. Moreover, we determined the transcriptional and morphological sequences of key developmental events associated with spinogenesis, including the conversion of drebrin to its brain-specific isoform A and the N-methyl-D-aspartate (NMDA) receptor subunit switch. TF-induced iPSC neurons successfully acquired structural and functional synaptic maturity, which will critically expand their utility in modeling higher brain functions and disorders.},
keywords = {Activity Scan Assay, MaxTwo, Network Assay},
pubstate = {published},
tppubtype = {article}
}
2022

title = {Downregulation of PMP22 ameliorates myelin defects in iPSC-derived human organoid cultures of CMT1A},
author = {Jonas Van Lent and Leen Vendredy and Elias Adriaenssens and Tatiana Da Silva Authier and Bob Asselbergh and Marcus Kaji and Sarah Weckhuysen and Ludo Van Den Bosch and Jonathan Baets and Vincent Timmerman},
url = {https://academic.oup.com/brain/advance-article/doi/10.1093/brain/awac475/6895197?login=false},
doi = {https://doi.org/10.1093/brain/awac475},
year = {2022},
date = {2022-12-12},
journal = {Brain},
abstract = {Charcot-Marie-Tooth (CMT) disease is the most common inherited disorder of the peripheral nervous system. CMT1A accounts for 40-50% of all cases and is caused by a duplication of the PMP22 gene on chromosome 17, leading to dysmyelination in the peripheral nervous system. Patient-derived models to study such myelination defects are lacking as the in vitro generation of human myelinating Schwann cells has proven to be particularly challenging. Here, we present an iPSC-derived organoid culture, containing various cell types of the peripheral nervous system, including myelinating human Schwann cells, which mimics the human peripheral nervous system. Single-cell analysis confirmed the peripheral nervous system-like cellular composition and provides insight into the developmental trajectory. We used this organoid-model to study disease signatures of CMT1A, revealing early ultrastructural myelin alterations, including increased myelin periodic line distance and hypermyelination of small axons. Furthermore, we observed the presence of onion bulb-like formations in a later developmental stage. These hallmarks were not present in the for CMT1A-corrected isogenic line or in a CMT2A iPSC line, supporting the notion that these alterations are specific to CMT1A. Downregulation of PMP22 expression using short-hairpin RNAs or a combinatorial drug consisting of baclofen, naltrexone hydrochloride and D-sorbitol, was able to ameliorate the myelin defects in CMT1A-organoids. In summary, this self-organizing organoid model is able to capture biologically meaningful features of the disease and capture the physiological complexity, forms an excellent model to study demyelinating diseases, and supports the therapeutic approach of reducing PMP22 expression.},
keywords = {Activity Scan Assay, Axon Tracking Assay, MaxTwo, Network Assay, Organoids},
pubstate = {published},
tppubtype = {article}
}
![]()
title = {A multi-modal fitting approach to construct single-neuron models with patch clamp and high-density microelectrode arrays},
author = {Buccino, Alessio Paolo; Damart, Tanguy; Bartram, Julian; Mandge, Darshan; Xue, Xiaohan; Zbili, Mickael; Gänswein, Tobias; Jaquier, Aurélien; Emmenegger, Vishalini; Markram, Henry; Hierlemann, Andreas; Van Geit, Werner.},
doi = {https://doi.org/10.1101/2022.08.03.502468},
year = {2022},
date = {2022-08-11},
journal = {bioRxiv},
abstract = {In computational neuroscience, multicompartment models are among the most biophysically realistic representations of single neurons. Constructing such models usually involves the use of the patch-clamp technique to record somatic voltage signals under different experimental conditions. The experimental data are then used to fit the many parameters of the model. While patching of the soma is currently the gold-standard approach to build multicompartment models, several studies have also evidenced a richness of dynamics in dendritic and axonal sections. Recording from the soma alone makes it hard to observe and correctly parameterize the activity of non-somatic compartments.
In order to provide a richer set of data as input to multicompartment models, we here investigate the combination of somatic patch-clamp recordings with recordings of high-density micro-electrode arrays (HD-MEAs). HD-MEAs enable the observation of extracellular potentials and neural activity of neuronal compartments at sub-cellular resolution.
In this work, we introduce a novel framework to combine patch-clamp and HD-MEA data to construct multicompartment models. We first validate our method on a ground-truth model with known parameters and show that the use of features extracted from extracellular signals, in addition to intracellular ones, yields models enabling better fits than using intracellular features alone. We also demonstrate our procedure using experimental data by constructing cell models from in vitro cell cultures.
The proposed multi-modal fitting procedure has the potential to augment the modeling efforts of the computational neuroscience community and to provide the field with neuronal models that are more realistic and can be better validated.
Author Summary Multicompartment models are one of the most biophysically detailed representations of single neurons. The vast majority of these models are built using experimental data from somatic recordings. However, neurons are much more than just their soma and one needs recordings from distal neurites to build an accurate model. In this article, we combine the patch-clamp technique with extracellular high-density microelectrode arrays (HD-MEAs) to compensate this shortcoming. In fact, HD-MEAs readouts allow one to record the neuronal signal in the entire axonal arbor. We show that the proposed multi-modal strategy is superior to the use of patch clamp alone using an existing model as ground-truth. Finally, we show an application of this strategy on experimental data from cultured neurons.},
keywords = {2D Neuronal Culture, Activity Scan Assay, Axon Tracking Assay, HD-MEA, MaxOne, Other Tissues, Publication, Stimulation Assay},
pubstate = {published},
tppubtype = {article}
}
In order to provide a richer set of data as input to multicompartment models, we here investigate the combination of somatic patch-clamp recordings with recordings of high-density micro-electrode arrays (HD-MEAs). HD-MEAs enable the observation of extracellular potentials and neural activity of neuronal compartments at sub-cellular resolution.
In this work, we introduce a novel framework to combine patch-clamp and HD-MEA data to construct multicompartment models. We first validate our method on a ground-truth model with known parameters and show that the use of features extracted from extracellular signals, in addition to intracellular ones, yields models enabling better fits than using intracellular features alone. We also demonstrate our procedure using experimental data by constructing cell models from in vitro cell cultures.
The proposed multi-modal fitting procedure has the potential to augment the modeling efforts of the computational neuroscience community and to provide the field with neuronal models that are more realistic and can be better validated.
Author Summary Multicompartment models are one of the most biophysically detailed representations of single neurons. The vast majority of these models are built using experimental data from somatic recordings. However, neurons are much more than just their soma and one needs recordings from distal neurites to build an accurate model. In this article, we combine the patch-clamp technique with extracellular high-density microelectrode arrays (HD-MEAs) to compensate this shortcoming. In fact, HD-MEAs readouts allow one to record the neuronal signal in the entire axonal arbor. We show that the proposed multi-modal strategy is superior to the use of patch clamp alone using an existing model as ground-truth. Finally, we show an application of this strategy on experimental data from cultured neurons.
2020

title = {MAPSYNE: Miniaturized micropipette system combined with high-density microelectrode arrays for automated manipulation of neuronal networks in-vitro},
author = {Chiara Ricci and Urs Frey and Marie Engelene J. Obien},
url = {https://ieeexplore.ieee.org/document/9175797/},
doi = {10.1109/EMBC44109.2020.9175797},
year = {2020},
date = {2020-10-08},
journal = {IEEE},
abstract = {We present MAPSYNE, a miniaturized and automated system combining a high-density microelectrode array (HD-MEA) and a movable micropipette for studying, monitoring, and perturbing neurons in vitro. The system involves an all-electrical approach to automatically move a glass micropipette towards a target location on the HD-MEA surface, without the need for a microscope. Two methods of performing blind navigation are employed, (i) stop-measure-go approach wherein the pipette moves for a predefined distance before measuring its location then the process is repeated until the pipette reaches its destination, and (ii) predictive approach wherein the pipette is continuously tracked and moved. This automated system can be applied for unsupervised single-cell manipulation of neurons in a network, such as electroporation and local delivery of compounds.},
keywords = {Activity Scan Assay, HD-MEA, MaxOne, MEA Technology},
pubstate = {published},
tppubtype = {article}
}